
Job Seeker Support
What is Jobvite?
Jobvite is a Talent Acquisition software suite and Applicant Tracking System (ATS) – online software that businesses use to organize their hiring process, curate employer branding, perform recruitment marketing, manage applications, and host their career pages.
Jobvite is not a job board, and, unfortunately, cannot help you get a job. If you’ve arrived to this page from a company’s open jobs career page seeking employment, please navigate back to the website where you originally tried to apply for a job and try again. If you end up back here, please reach out to the organization you’re interested in working for directly. Most of our customers broadcast open roles on all major job boards like Indeed, LinkedIn, ZipRecruiter, Glassdoor, and use employer-branded career pages, “Powered by Jobvite”. Please do not fill out Jobvite demo or contact forms to submit employment inquiry, and instead visit those sites directly to search or ask about open roles. We wish you luck during your search!
If you are interested in a career with Jobvite (an Employ, Inc. company), please take a look at our open positions.
If you are interested in purchasing a Jobvite solution for your company, please contact us.
For job seekers actively using the Jobvite platform to apply for roles and opportunities, you may find the resources below helpful to you.
Job Seeker Resources:
Jobvite Login Issues
How do I submit a support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.
I forgot my password
Go to Jobvite.com, click on “Job Seeker Login,” add your email address, and then click on “Forgot Password.” You will then receive an email with a link to a page that will prompt you to ‘Enter’ and ‘Confirm’ your new password.
Application Issues
What does my “job application status” mean in Jobvite?
- New: Application received.
- In Process: The company is reviewing the application.
- Not Selected: The company has not selected the applicant for the position.
- Hired: The candidate has been selected for the position.
- Closed: The job has been closed. No further information is available.
Some companies may use application statuses that are different from this default list. To learn more about your application status, please contact the company you applied to directly. Jobvite does not change or have any influence on the status of an application.
How do I apply for a job from my job seeker account?
You cannot submit applications/resumes through your Job Seeker account. If a company requests that you submit the initial application and resume for a position, you will need to visit their career site or any other Job Board link or posting that is available to you.
How do I check the status of my resume through Jobvite?
After you applied, you should have received a confirmation email that the company received your application. If there’s a link in the email, then please follow that link to check your status. If not, you’ll need to contact the company that you are applying to directly.
When I submit my resume to Jobvite, I get an error or notice that says, “You have already applied to the job.” Why?
With rare exceptions, you cannot apply to the same job twice. You can access your previous application through your job seeker account to contact the hiring team if you would like to request being reconsidered.
After I have submitted my application, can Jobvite help me get hired at a company I want to work for?
Jobvite is recruiting software used by thousands of companies to manage their recruiting process and plays no part in hiring decisions.
How do I submit a support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.
Resume and LinkedIn Issues
Why am I having issues uploading my resume on the apply page?
Currently, you cannot add resumes that are scanned or locked PDFs. Please be sure that the document is a Word document or unlocked PDF. After uploading your resume to populate application fields, you may still need to use the Attach button to include your resume as part of your application file.
Why does my resume lose my formatting after I’ve uploaded it?
What you are seeing is a text only preview of your resume. The parsing is for internal search purposes and the recruiter will receive a copy of your resume in the original format (.PDF, .DOC, etc.) as well.
How do I update my resume with a company I have applied to?
Reply to the confirmation email you get with the new resume. Be sure to let the company know that you are re-submitting with an updated resume.
How do I update my LinkedIn profile on Jobvite?
If you have included your LinkedIn profile in your job application, it cannot be removed or edited within your application page. Essentially, when you add your LinkedIn profile to a job application, you are adding a link to your current LinkedIn profile. The profile will always show your most recent updates when viewed by the hiring team. So, make any desired changes or updates directly within LinkedIn.
How do I submit a support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.
Email Issues
Why did I not receive a confirmation email for my application?
Typically, confirmation emails are sent right away, though sometimes prospective employers have email settings in place that will cause a slight delay. If some time has gone by and you have not received a confirmation message, then please check your Spam or Junk folders.
Can I reply directly to confirmation emails or other messages I receive? If so, who receives my reply?
You can reply directly to the application confirmation email and your reply will be sent to the recruiter who is managing the job posting you have applied to.
Why are my emails from Jobvite being sent to my spam folder?
Jobvite emails come from no-reply@jobvite-inc.com. Please add this email to your safe sender list. Password reset emails are also sent from no-reply@jobvite-inc.com.
How do I stop getting notifications?
When you click on any link from a Jobvite email, there’s a menu item called ‘Change Notifications.’ When you’re logged into Jobvite, go to https://hire.jobvite.com/Jobs/Notify.aspx, then to Preferences to change your notification settings.
How do I change my email address with Jobvite?
To update your email address, you will want to login to your Jobvite account and select the application you are looking to update. From here, use the ‘New Message’ option to send a message to the recruiter with your updated email address.
How do I submit a support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.
Video Issues
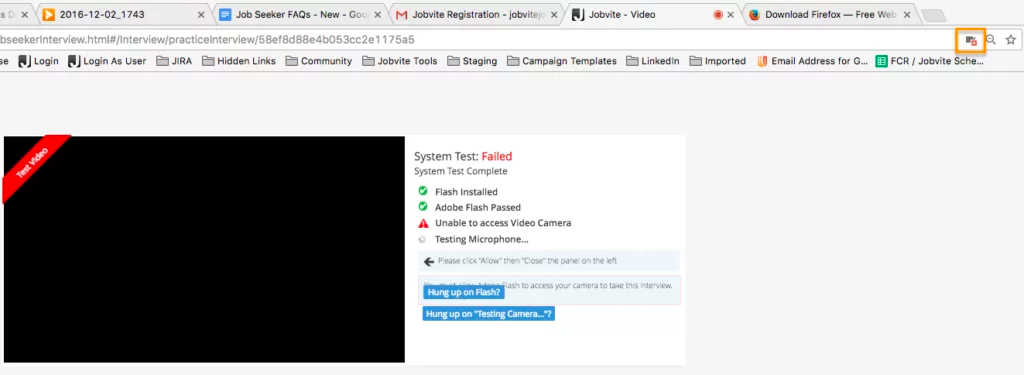
I’m having a problem with the camera and/or microphone access for a video interview. Why?
Please make sure your web browser is set to allow video and microphone access, or the camera and microphone icons will continue to spin when performing the System Test. Also make sure that you’re using the most up-to-date browser.
In Google Chrome, there will be a camera icon in the URL search bar. Please click the icon and make sure the camera and microphone you need to access for your interview is selected and access permission is accepted.
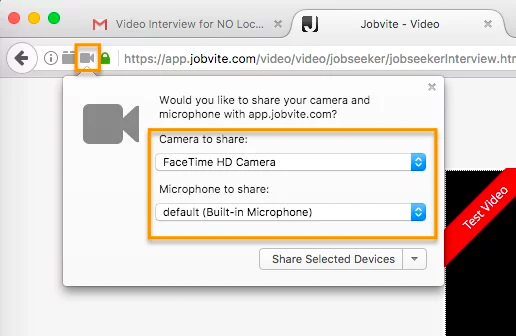
In Firefox, there’s a lock icon to the left of the URL window which will allow you to give permission to the camera and microphone.
How do I submit a Support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.
Delete Info and Account
How do I get my personal information removed from a company I have applied to?
Due to our privacy policy and contracts with the companies using our software, you’ll need to contact the company you’ve applied to delete your personal information.
How do I delete my application?
An application cannot be deleted after it has been submitted, but you can retract your application by contacting the company you applied to directly.
How do I delete my Jobvite account?
First, it’s important to know that deleting your Jobvite account does not remove any applications from the companies you previously applied to. We can’t remove those applications because they are required to track all candidate applications for compliance reasons. Therefore, it probably isn’t necessary to delete your account. If you would still like to delete your account, then please click here to submit a Job Seeker Support Case and complete all the required fields.
How do I submit a support case with Jobvite?
Click here to submit a Job Seeker Support Case and complete all the required fields.